class: center, middle, inverse, title-slide # Unsupervised learning: continuous latent variable models ## Foundational Computational Biology II ### Kieran Campbell ### Lunenfeld Tanenbaum Research Institute & University of Toronto ### 2021-02-22 (updated: 2022-04-04) --- class: inverse, middle # What we'll cover 1. A historical perspective on continuous latent variable models 2. Principal component analysis and pPCA 3. Non-negative matrix factorization 4. Nonlinear methods: tSNE & autoencoders --- # Conventions * We'll have `\(N\)` samples indexed `\(n = 1, \ldots, N\)` -- * When an output is multidimensional, we'll refer to output dimensions as `\(p=1,\ldots,P\)` -- * We define a _latent variable_ as a sample specific unobserved variable * e.g. "What cluster does sample `\(n\)` belong to?" -- * Latent variables are `\(z_n\)` (possibly high dimensional, possibly discrete) -- * Observed variables are `\(y_n\)` --- # Why use continuous latent variables? Many high-dimensional data are generated from an intrinsically lower-dimensional space ("manifold") -- ## Examples: * Imaging data: images have lower intrinsict degrees of freedom than length `\(\times\)` height pixels .center[ <img src="unsupervised-learning-figs/three.png" width=500> ] .footnote[ Figure credit: [Bis06] ] * Single-cell transcriptomic data --- # Origins of continuous latent variable models Spearman [Spe04] noted in 1904 correlation between pupil's school grades: | Student | Maths | English | Science | | -- | -- | -- | -- | | Alice | 68 | 79 | 65 | | Bob | 56 | 47 | 60 | | Carol | 92 | 85 | 84 | | ... | ... | ... | ... | -- > What if the grades aren't just correlated with each other, but depend on an unseen ("latent") variable...called _general intelligence_ ? --- # Origins of continuous latent variable models How would we describe this mathematically? -- * `\(y_{np}\)` grade of pupil `\(n\)` in subject `\(p\)` * `\(z_n\)` "intelligence" pupil `\(n\)` * `\(w_{p}\)` describes how the intelligence impacts the grade -- Then `$$y_{np} = w_p z_n + \epsilon_{np}$$` where `\(\epsilon_{np}\)` is the error term -- This (vastly oversimplified) model is not a good model of reality. More reading: https://www.stat.cmu.edu/~cshalizi/402/lectures/18-factors/lecture-18.pdf --- # Modern applications Where do such models appear today? -- Anywhere with high dimensional data, especially genomics -- ## Single-cell gene expression data * `\(y_{np}\)` expression gene `\(p\)` in cell `\(n\)` * `\(z_n\)` cell cycle progression of cell `\(n\)` * `\(w_p\)` how cell cycle influences gene `\(p\)` `$$y_{np} = w_p z_n + \epsilon_{np}$$` -- .center[ <img src="intro-ml_figs/3x/pseudotime-example@3x.png" width=500> ] --- # Latent variable models as matrix factorization Suppose we model `\(K\)` latent variables rather than just 1 `$$y_{np} = \sum_{k=1}^K w_{pk} z_{nk} + \epsilon_{np}$$` -- In matrix form: $$ `\begin{aligned} \mathbf{Y} = \mathbf{Z} \mathbf{W} + \mathbf{E} \end{aligned}` $$ -- 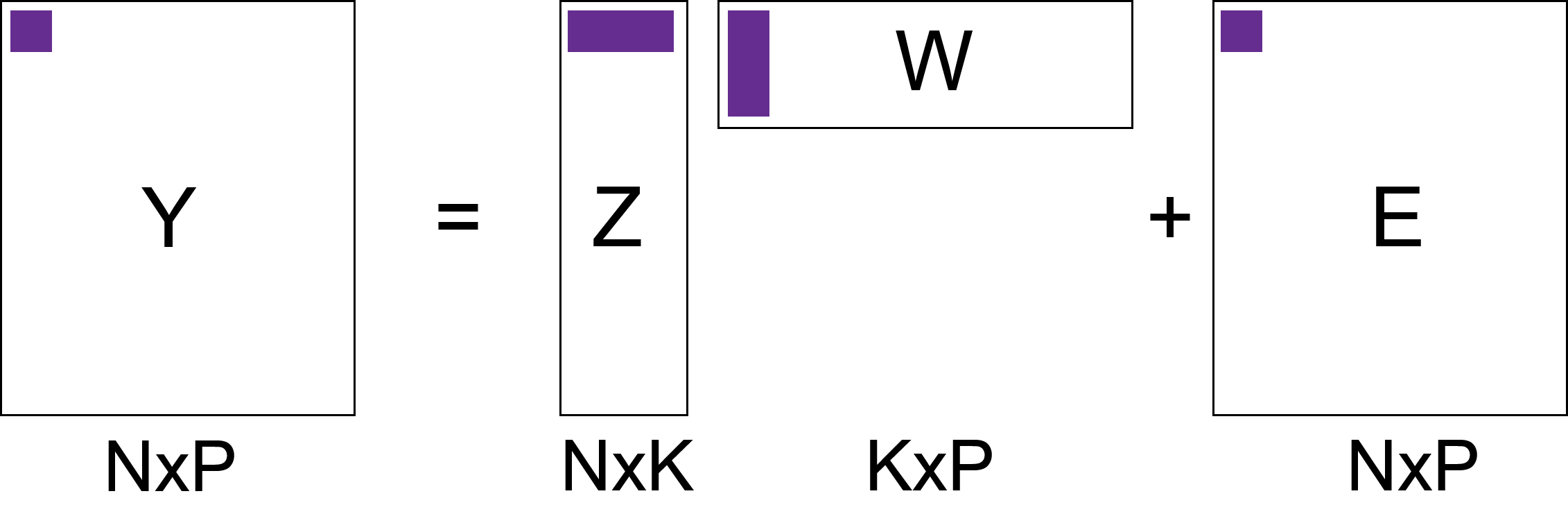 --- # Some notes .center[ <img src="intro-ml_figs/3x/matrix-factorization@3x.png" width=400> ] -- * Dimensionality reduction: for each `\(n\)` we find a lower dimensional representation (length `\(K\)` rather than `\(P\)`) -- * Also (probabilistic) matrix factorization -- * We've said nothing about `\(Y\)`, `\(Z\)`, `\(W\)`, `\(E\)`. Depending on these: * PCA, probabilistic PCA * Factor analysis * Non-negative matrix factorization --- # Principal component analysis Aims to project data onto a space `\(K < P\)` such that -- * Each of the `\(K\)` components are orthogonal -- * The projection of points onto the first component retains the most variance -- * Projection onto second retains second most variance -- * ... --- # Principal component analysis visualized .pull-left[ * `\(P=2\)`, `\(K=1\)` * White dots are observed data * Green line is the subspace (principal component) * Blue dots are projections onto the principal component ] .pull-right[ .center[ <img src="unsupervised-learning-figs/pca_figure1.jpg" width=400> ] ] .footnote[ Image credit: Lior Pachter https://liorpachter.wordpress.com/2014/05/26/what-is-principal-component-analysis/ ] --- # Differences from linear regression .center[ <img src="unsupervised-learning-figs/pca_figure2.jpg" width=400> ] .footnote[ Image credit: Lior Pachter https://liorpachter.wordpress.com/2014/05/26/what-is-principal-component-analysis/ ] --- # Calculation of principal components Let `\(\mathbf{u}_1 \in \mathbb{R^P}\)` project each data point onto the first principal component: `$$z_{n1} = \mathbf{u}_1^\mathrm{T} \mathbf{y}_n$$` such that `\(\mathbf{u}_1^\mathrm{T} \mathbf{u}_1 = 1\)` -- Then as shown in Bishop [Bis06], to find `\(\mathbf{u}_1\)` we solve the eigenvalue equation `$$S\mathbf{u}_1 = \lambda \mathbf{u}_1$$` -- Where `\(S\)` is the `\(P \times P\)` empirical covariance `$$S = \frac{1}{N} \sum_{n=1}^N (\mathbf{y_n} - \bar{\mathbf{y}}) (\mathbf{y_n} - \bar{\mathbf{y}})^\mathrm{T}$$` -- `\(\mathbf{u}_1\)` is the eigenvalue corresponding to the largest eigenvalue `\(\lambda\)`, `\(\mathbf{u}_2\)` second largest, etc. --- # An penguin example |species | bill_length_mm| bill_depth_mm| flipper_length_mm| body_mass_g| |:-------|--------------:|-------------:|-----------------:|-----------:| |Adelie | 39.1| 18.7| 181| 3750| |Adelie | 39.5| 17.4| 186| 3800| |Adelie | 40.3| 18.0| 195| 3250| |Adelie | 36.7| 19.3| 193| 3450| -- <img src="22-unsupervised_learning_continuous_files/figure-html/unnamed-chunk-3-1.png" style="display: block; margin: auto;" /> --- # An penguin example (II) |species | bill_length_mm| bill_depth_mm| flipper_length_mm| body_mass_g| |:-------|--------------:|-------------:|-----------------:|-----------:| |Adelie | 39.1| 18.7| 181| 3750| |Adelie | 39.5| 17.4| 186| 3800| |Adelie | 40.3| 18.0| 195| 3250| |Adelie | 36.7| 19.3| 193| 3450| * `\(P\)` is {bill_length_mm, bill_depth_mm, flipper_length_mm, body_mass_g} * `\(N\)` is each row (each individual penguin measured) --- # PCA on Penguins ```r peng_mat <- scale(as.matrix(penguins[,2:5])) pca <- prcomp(peng_mat) ``` -- We can look at `\(\mathbf{u}_1, \ldots, \mathbf{u}_4\)`: ```r pca$rotation ``` ``` ## PC1 PC2 PC3 PC4 ## bill_length_mm 0.4537532 -0.60019490 -0.6424951 0.1451695 ## bill_depth_mm -0.3990472 -0.79616951 0.4258004 -0.1599044 ## flipper_length_mm 0.5768250 -0.00578817 0.2360952 -0.7819837 ## body_mass_g 0.5496747 -0.07646366 0.5917374 0.5846861 ``` -- And the projections `\(\mathbf{z}_1\)`: ```r head(pca$x, n=1) ``` ``` ## PC1 PC2 PC3 PC4 ## [1,] -1.850808 -0.03202119 0.2345487 0.5276026 ``` --- # Calculating the projections Recall `\(\mathbf{z}_{1} = \mathbf{u}_1^\mathrm{T} \mathbf{y}\)`: ```r t(pca$rotation[,1,drop=FALSE]) %*% peng_mat[1,] ``` ``` ## [,1] ## PC1 -1.850808 ``` and verify this is what's calculated by `prcomp`: ```r pca$x[1,1] ``` ``` ## PC1 ## -1.850808 ``` --- # Graphing the results ```r df <- as_tibble(pca$x) %>% mutate(species = penguins$species) ggplot(df, aes(x = PC1, y = PC2, colour = species)) + geom_point() ``` <img src="22-unsupervised_learning_continuous_files/figure-html/unnamed-chunk-10-1.png" style="display: block; margin: auto;" /> --- # Probabilistic PCA Variant of PCA called probabilistic PCA (pPCA) .center[ <img src="intro-ml_figs/3x/matrix-factorization@3x.png" width=400> ] specified by the following probabilistic model: `$$\mathbf{z}_n \sim \mathcal{N}(0,1)$$` $$\mathbf{y}_n = \mathcal{N}(\mathbf{W}^\mathrm{T}\mathbf{z}_n, \sigma^2 \mathbf{I}) $$ -- * Can derive a computationally-efficient EM algorithm that avoids computing full covariance matrix * Allows for missing data * Building block for more interesting probabilistic models --- # Visual representation of pPCA .center[ <img src="unsupervised-learning-figs/pca_figure3.jpg" width=400> ] .footnote[ Image credit: Lior Pachter https://liorpachter.wordpress.com/2014/05/26/what-is-principal-component-analysis/ ] --- # The rotation problem Remember `\(\mathbf{y}_n \sim \mathcal{N}(\mathbf{W}^\mathrm{T}\mathbf{z}_n, \sigma^2 \mathbf{I})\)` where `\(\mathbf{W}\)` is `\(K \times P\)` and `\(\mathbf{z}_n\)` is `\(K \times 1\)` -- We can introduce a `\(K \times K\)` rotation matrix `\(R\)` s.t. `\(RR^\mathrm{T}=\mathbf{I}\)` and get $$ `\begin{aligned} \mathbf{W}^\mathrm{T}\mathbf{z}_n & = \mathbf{W}^\mathrm{T}\mathbf{I} \mathbf{z}_n \\ & = \mathbf{W}^\mathrm{T} R R^\mathrm{T} \mathbf{z}_n \\ & = (R^\mathrm{T} \mathbf{W})^\mathrm{T} R^\mathrm{T} \mathbf{z}_n \\ & =\tilde{\mathbf{W}}^\mathrm{T} \tilde{\mathbf{z}}_n \end{aligned}` $$ -- In other words we can get _exactly_ the same likelihood for a rotated solution `\(\tilde{\mathbf{W}}\)`, `\(\tilde{\mathbf{z}}_n\)` ! --- # Non-negative matrix factorization Paatero and Tapper [PT94] introduced NMF .center[ <img src="intro-ml_figs/3x/matrix-factorization@3x.png" width=400> ] `\(\mathbf{Y}, \mathbf{Z}, \mathbf{W}\)` are _all non-negative_ -- Solutions found via minimizing `$$\sum_{n=1}^N \sum_{p=1}^P \left(\mathbf{Y} - \mathbf{Z}\mathbf{W} \right)_{np}^2$$` -- This is a hard, _non unique_ optimization problem Nice discussion: https://blog.acolyer.org/2019/02/18/the-why-and-how-of-nonnegative-matrix-factorization/ --- # Applications of NMF Very popular in computational biology -- * Data `\(\mathbf{Y}\)` is often non-negative (counts, intensities) -- * `\(\mathbf{W}\)` being non-negative provides a "contribution" from each component `\(K\)` -- * If `\(z_{nk} = 0\)`, component `\(k\)` is _inactive_ in sample `\(n\)` -- * If `\(z_{nk}\)` is large, component `\(k\)` is _active_ in sample `\(n\)` --- # Example: mutational signatures How do we find common patterns of mutation along the genome? -- Think of all _trinucleotide_ substitutions, e.g. `ACG` <svg viewBox="0 0 448 512" xmlns="http://www.w3.org/2000/svg" style="height:1em;fill:currentColor;position:relative;display:inline-block;top:.1em;"> [ comment ] <path d="M190.5 66.9l22.2-22.2c9.4-9.4 24.6-9.4 33.9 0L441 239c9.4 9.4 9.4 24.6 0 33.9L246.6 467.3c-9.4 9.4-24.6 9.4-33.9 0l-22.2-22.2c-9.5-9.5-9.3-25 .4-34.3L311.4 296H24c-13.3 0-24-10.7-24-24v-32c0-13.3 10.7-24 24-24h287.4L190.9 101.2c-9.8-9.3-10-24.8-.4-34.3z"></path></svg> `AAG` -- There are `\(96\)` possible mutations <svg viewBox="0 0 448 512" xmlns="http://www.w3.org/2000/svg" style="height:1em;fill:currentColor;position:relative;display:inline-block;top:.1em;"> [ comment ] <path d="M190.5 66.9l22.2-22.2c9.4-9.4 24.6-9.4 33.9 0L441 239c9.4 9.4 9.4 24.6 0 33.9L246.6 467.3c-9.4 9.4-24.6 9.4-33.9 0l-22.2-22.2c-9.5-9.5-9.3-25 .4-34.3L311.4 296H24c-13.3 0-24-10.7-24-24v-32c0-13.3 10.7-24 24-24h287.4L190.9 101.2c-9.8-9.3-10-24.8-.4-34.3z"></path></svg> `\(N \times 96\)` count matrix -- Apply NMF to this matrix: .center[ <img src="unsupervised-learning-figs/mut-sig-nmf.png" width=600> ] --- # Example: mutational signatures (II) .pull-left[ Each of the `\(K\)` components is known as a _mutational signature_ ] .pull-right[ .center[ <img src="unsupervised-learning-figs/mut-sig-nmf.png" width=300> ] ] -- Each tumour sample is then defined by activation of multiple mutational signatures (via `\(\mathbf{Z}\)`) -- Each mutational signature is defined by contributions from each mutation type .center[ <img src="unsupervised-learning-figs/SBS4.png" width=400> ] .footnote[ Figure: Alexandrov, Kim, Haradhvala, Huang, Ng, Wu, Boot, Covington, Gordenin, Bergstrom, and others [Ale+20] ] Often associated with known mutational processes, e.g. double strand break repair, UV light exposure, etc. --- # Nonlinear dimensionality reduction So far all mappings have been linear: * `\(z\)` <svg viewBox="0 0 448 512" xmlns="http://www.w3.org/2000/svg" style="height:1em;fill:currentColor;position:relative;display:inline-block;top:.1em;"> [ comment ] <path d="M190.5 66.9l22.2-22.2c9.4-9.4 24.6-9.4 33.9 0L441 239c9.4 9.4 9.4 24.6 0 33.9L246.6 467.3c-9.4 9.4-24.6 9.4-33.9 0l-22.2-22.2c-9.5-9.5-9.3-25 .4-34.3L311.4 296H24c-13.3 0-24-10.7-24-24v-32c0-13.3 10.7-24 24-24h287.4L190.9 101.2c-9.8-9.3-10-24.8-.4-34.3z"></path></svg> `\(y\)` via `\(\mathbf{y} = \mathbf{W}^\mathrm{T} \mathbf{z}\)` (e.g. pPCA, NMF) * `\(y\)` <svg viewBox="0 0 448 512" xmlns="http://www.w3.org/2000/svg" style="height:1em;fill:currentColor;position:relative;display:inline-block;top:.1em;"> [ comment ] <path d="M190.5 66.9l22.2-22.2c9.4-9.4 24.6-9.4 33.9 0L441 239c9.4 9.4 9.4 24.6 0 33.9L246.6 467.3c-9.4 9.4-24.6 9.4-33.9 0l-22.2-22.2c-9.5-9.5-9.3-25 .4-34.3L311.4 296H24c-13.3 0-24-10.7-24-24v-32c0-13.3 10.7-24 24-24h287.4L190.9 101.2c-9.8-9.3-10-24.8-.4-34.3z"></path></svg> `\(z\)` via `\(z = \mathbf{u}^\mathrm{T} \mathbf{y}\)` (PCA) but we may wish to learn more flexible, nonlinear embeddings that capture more complex structure -- Let's look at 1. tSNE 2. Autoencoders --- # t-distributed stochastic neighbour embedding (tSNE) <!-- Compute "conditional neighbour probabilities" --> <!-- `$$p_{j|i} = \frac{\exp(-||\mathbf{y}_i - \mathbf{y}_j||^2/2\sigma_i^2)}{\sum_{k\neq i} \exp(-||\mathbf{y}_i - \mathbf{y}_k ||^2/2\sigma_i^2)}$$` --> <!-- and symmetrize to make `\(p_{ij} = (p_{j|i} + p_{i|j})/2N\)` --> <!-- then introduce --> <!-- $$q_{ij} = $$ --> tSNE [VH08] is a method for learning non-linear reduced-dimension representations .pull-left[ * No explicit forward or reverse map * _Perplexity_ parameter controls crowdedness of representation * Reproduces local similarity more faithfully than global similarity * _Very_ popular in single-cell transcriptomics **How to Use t-SNE Effectively**: https://distill.pub/2016/misread-tsne/ ] .pull-right[ .center[ <img src="unsupervised-learning-figs/tabulamuris.png" width=400> ] ] .footnote[ Image: [C18] ] --- # Autoencoders One approach is to 1. Map `\(y\)` to `\(z\)` using a deep neural network (the _encoder_) -- 2. Map `\(z\)` to a reconstructed `\(\hat{y}\)` using a second deep neural network (the _decoder_) -- 3. Adjust the parameters of both networks to minimize `\((y - \hat{y})^2\)` --- # Deep neural networks Let `\(\mathbf{x} = x_1, x_2, x_3\)` be the inputs -- (feed forward) neural networks reach an output by iteratively applying: * Linear layer: `\(\sum_i w_i x_i + b\)` * A non-linearity such as sigmoid, tanh to map from input `\(\mathbf{x}\)` to output `\(y\)` -- .center[ <img src="unsupervised-learning-figs/deep neural networks.png" width=500> ] --- # Autoencoders Introduce _encoder_ network parameterized by `\(\theta\)`: `$$\mathbf{z} = \texttt{enc}_\theta(\mathbf{y})$$` -- _Decoder_ to map back to predicted values `\(\hat{\mathbf{y}}\)`: `$$\hat{\mathbf{y}} = \texttt{dec}_\phi(\mathbf{z}) = \texttt{dec}_\phi(\texttt{enc}_\theta(\mathbf{y}))$$` -- Then update `\(\phi\)`, `\(\theta\)` to minimize `$$(\mathbf{y} - \hat{\mathbf{y}}^2) = (\mathbf{y} - \texttt{dec}_\phi(\texttt{enc}_\theta(\mathbf{y})))^2$$` -- Since `\(\mathbf{z}\)` is of size `\(K < P\)` this performs nonlinear dimensionality reduction -- Becoming very popular in single-cell RNA-sequencing, see e.g. [Lop+18] --- # References These slides: [camlab.ca/teaching](https://www.camlab.ca/teaching) Alexandrov, L. B., J. Kim, N. J. Haradhvala, et al. (2020). "The repertoire of mutational signatures in human cancer". In: _Nature_ 578.7793, pp. 94-101. Bishop, C. M. (2006). _Pattern recognition and machine learning_. springer. Consortium, T. M. and others (2018). "Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris". In: _Nature_ 562.7727, pp. 367-372. Lopez, R., J. Regier, M. B. Cole, et al. (2018). "Deep generative modeling for single-cell transcriptomics". In: _Nature methods_ 15.12, pp. 1053-1058. Paatero, P. and U. Tapper (1994). "Positive matrix factorization: A non-negative factor model with optimal utilization of error estimates of data values". In: _Environmetrics_ 5.2, pp. 111-126. Spearman, C. (1904). "" General Intelligence" Objectively Determined and Measured." Van der Maaten, L. and G. Hinton (2008). "Visualizing data using t-SNE." In: _Journal of machine learning research_ 9.11.